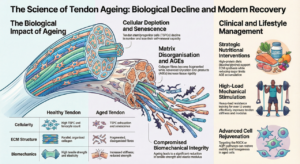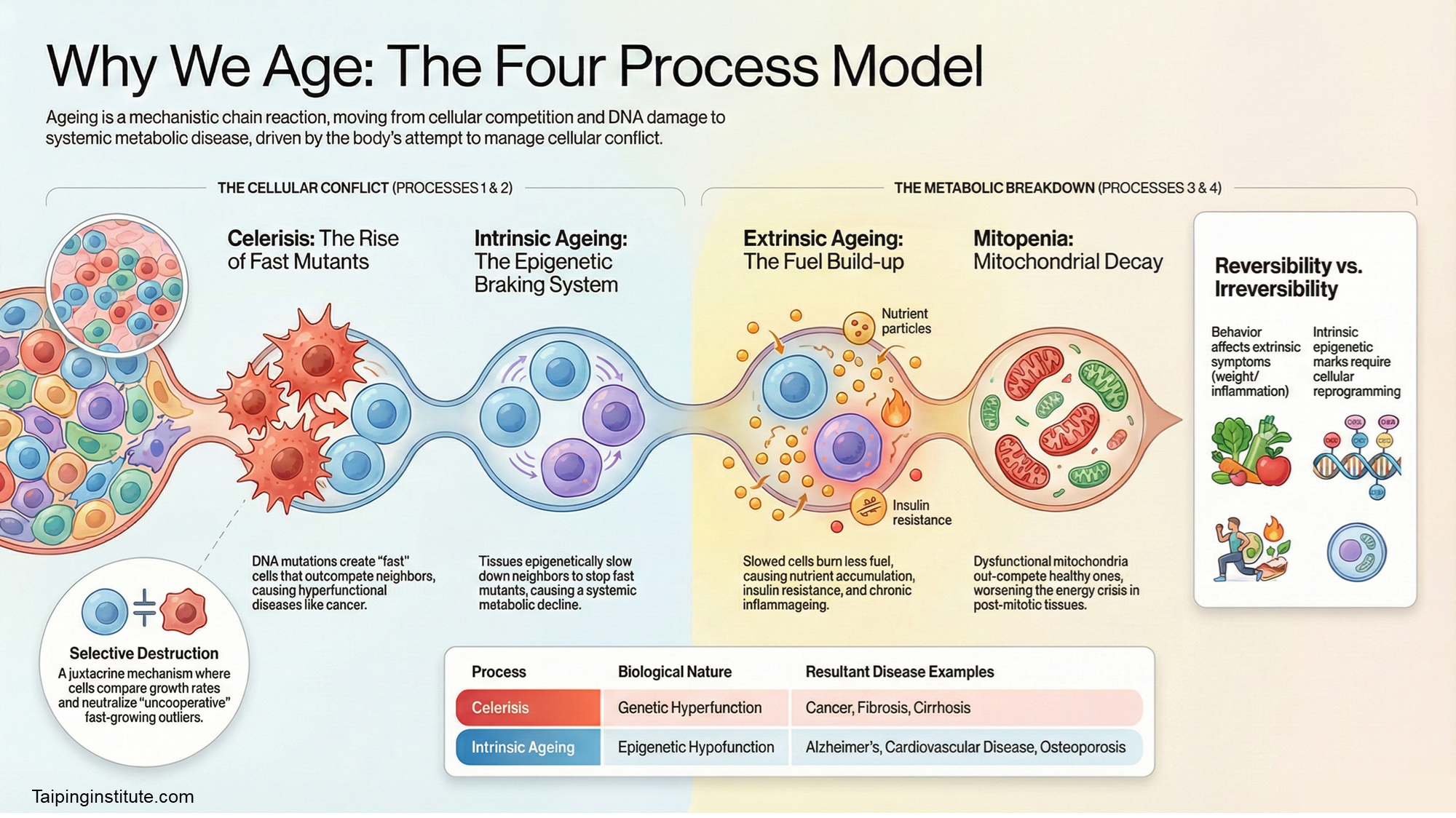
For decades, the standard scientific explanation for ageing has been almost apologetically vague: it is a “complex, multifactorial process” involving a slow accumulation of cellular errors. This view suggests that our bodies simply wear out, like old machinery, due to unavoidable DNA damage and toxic by-products. However, a unifying new “four process model” suggests that ageing is not a random slide into chaos, but a structured sequence of biological events driven by the forces of natural selection occurring within our own tissues. By viewing ageing through the lens of how cells compete and communicate, we can finally connect various “hallmarks” of decay, from cancer to weight gain, into a single, mechanistic map.
The first of these processes, dubbed celerisis, stems from a simple rule of biology: cells that proliferate and metabolise faster will naturally outcompete their slower neighbours. When DNA damage gives a cell a growth advantage, it spreads through the tissue like an invasive species. While this is evolutionarily efficient at a cellular level, for the organism it is disastrous, leading directly to “hyperfunctional” diseases such as cancer and fibrosis. Our tissues are essentially a battleground where the most aggressive cells strive to take over, threatening the stability of the entire body.
To prevent this internal takeover, our bodies have evolved a counter-measure known as intrinsic ageing. This is an active defence mechanism where slower-metabolising cells communicate with their faster neighbours via Notch signalling, using epigenetic “marks” to force them to slow down. Essentially, our tissues deliberately hit the brakes on growth to prevent cancer. The cost of this safety, however, is a progressive, body-wide metabolic slowdown. We do not age because our parts break; we age because our cells are forced to become sluggish to keep more dangerous mutants at bay.
This cellular slowdown triggers a third process: extrinsic ageing. As cells reduce their “anabolism” (growth and repair), they require less energy and therefore burn less fuel. Because the body maintains a strict equilibrium of bioavailable energy (ATP), this reduced demand causes mitochondria to shut down their furnaces. The unused glucose and lipids then begin to back up in the system, much like a factory with a stalled assembly line. This build-up induces insulin resistance as cells try to block further fuel entry, eventually leading to weight gain, metabolic syndrome, and the chronic “inflammageing” that defines late life.
In tissues where cells do not divide, such as the brain and heart, a fourth process occurs: mitopenia. Here, selection happens at the level of the mitochondria rather than the cell itself. Mutations in mitochondrial DNA can create organelles that are less functional but survive longer because they produce fewer reactive oxygen species (ROS). Over time, these “slower” but more persistent mitochondria dominate the cell, leading to the profound energy failures associated with Alzheimer’s and cardiovascular disease. This internal “survival of the slowest” mirrors the broader slowdown occurring across the rest of the body.
Understanding ageing as a sequence of evolved processes rather than a series of accidents changes how we might treat it. Current therapies like rapamycin or calorie restriction may work by mimicking or slowing these selection processes, while emerging tools like Yamanaka factors offer the potential to “reset” the epigenetic software of slowed cells. If ageing is indeed a shifted equilibrium designed to protect us from hyperactive disease, then the goal of future medicine will not just be to repair damage, but to safely retune the body’s metabolic pace back to the vigour of youth.